|
1. 1. KONFORMACIJSKA ANALIZA
Konformacijska analiza je samostalna i interdisciplinarna grana
kemije, stara jedva tridesetak godina, koja se bavi istraživanjem
strukturnih i energijskih svojstava molekule. Razvojem eksperimentalnih
(prije svega spektroskopskih) i teorijskih metoda (molekulske mehanike
i kvantne kemije), te posebice pojavom elektroničkih računala, konformacijska
analiza dobiva novi zamah. Naime, ona prestaje biti tek pukom "stereokemijom
rotacije oko jednostruke veze", već prerasta u samostalnu znanstvenu
disciplinu koja promatra najamanje promjene molekulske geometrije
i energije.(1)
1. 2. KONFORMACIJSKI IZOMERI
Konformacija se najlakše može definirati kao oblik koji molekula
poprima u prostoru uslijed rotacije oko jednostruke veze. Rotacijom
pojedinih dijelova molekule oko jednostrukih veza, atomi te molekule
mogu zauzimati različite položaje u prostoru, pri čemu se ne kidaju
veze među atomima u molekuli. Takvi različiti oblici molekula koji
nastaju okretanjem oko jednostrukih veza i koji lako prelaze jedan
u drugi zovu se konformacije. Najstabilnija konformacija(e) nekoga
spoja naziva se konformacijski izomer, konformer ili rotamer. Prema
tome, pojam konformera posljedica je unutrašnje molekulske rotacije.
(1,2) Razlika u rasporedu atoma između pojedinih konformera uzrokom
je i razlike u nizu njihovih fizičkih svojstava; potencijalna energija
molekule, dipolni moment, moment inercije, termodinamičke funkcije
itd. Također je poznato da se konformacijski izomeri, za razliku
od geometrijskih izomera, ne mogu izolirati i odvojiti, jer oni
mogu slobodno prelaziti iz jednoga izomera u drugi. Energijske barijere
konformacijskih prijelaza određene su geometrijskim značajkama molekule
i one prosječno iznose, u jednostavnim organskim molekulama, oko
20-25 kJmol-1. Dakle, da bi molekula prešla tu energijsku barijeru
potrebna joj je dovoljno velika unutrašnja energija. Pri sobnoj
temperaturi (25°C) unutrašnja energija molekule iznosi prosječno
oko 60-80 kJmol-1, što je dovoljno za prevladavanje energijske barijere
i brzu uzajamnu pretvrobu mogućih konformacijskih izomera. (1,2)
1. 3. KONFORMACIJSKA SVOJSTVA CIKLOHEKSANA
Cikloheksan je zasićeni ciklički ugljikovodik sa šest atoma ugljika
povezanih u prsten. Za razliku od planarne molekule benzena molekula
cikloheksana je neplanarne strukture. U slučaju planarnosti molekule
cikloheksana vezni kutovi C-C-C iznosili bi 120°, što je znatno
više od termodinamički najpovoljnijega tetraedarskog kuta (109.47°).
Uvijanjem planarnoga prstena vezni kutovi C-C-C se doista smanjuju
na 109.47°, rezultirajući termodinamički stabilnom neplanarnom strukturom
cikloheksana. Molekula cikloheksana može postojati u tri osnovne
neplanarne konformacije:
- konformacija stolice (engl. chair)
- konformacija kolijevke (engl. boat)
- konformacija izvijene kolijevke (engl twist-boat)
Termodinamički najpovoljnija konformacija cikloheksana jest konformacija
stolice (Slika 1.). Svi kutovi su tetraedarski, pa nema kutne napetosti.
U toj je konformaciji torzijska napetost minimalna, jer su sve skupine
i vodikovi atomi u nezasjenjenom, zvjezdastom položaju (engl. staggered).
Atomi vodika se nalaze na najvećoj mogućoj međusobnoj udaljenosti.
(1,2,3,4)
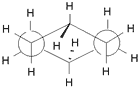
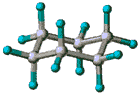
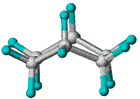
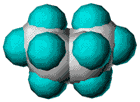
| Slika
1.
Prikazi molekule cikloheksana u konformaciji stolice. (a) perspektivni
prikaz, (b) Newman-ova projekcija, (c) molekulski modeli ("ball
and stick" i kalotni model). |
U konformaciji kolijevke (Slika 2.), također, nema kutne napetosti,
ali postoji znatna torzijska napetost. Sve skupine i vodikovi atomi
zauzimaju termodinamički nestabilniji zasjenjeni položaj (engl.
eclipsed). Nepovoljno nevezno međudjelovanje između dvaju vodikovih
atoma na suprotnim položajima prstena posljedica je njihove udaljenosti
od samo 1.83 A, što je manje od sume njihovih van der Waals-ovih
radijusa (2.00 A). Energija konformacije kolijevke je za oko 27
kJmol-1 viša od energije konformacije stolice pri 25°C (Slika 4.).
Posljedica energijskih razlika jest 99.9%-tna zastupljenost cikloheksana
u konformaciji stolice. (1,2,3,4)
| |
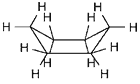
| Slika
2.
Prikazi molekule cikloheksana u konformaciji kolijevke. (a)
perspektivni prikaz, (b) Newman-ova projekcija, (c) molekulski
modeli ("ball and stick" i kalotni model). |
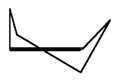
U konformaciji izvijene kolijevke (Slika 3.) smanjuje se, u usporedbi
s kolijevkom, torzijsko i nevezno međudjelovanje. Oblik kolijevke
se lagano izvija, pri čemu se atomi vodika odmiču svaki na drugu
stranu molekule, što smanjuje njihovo nevezno odbijanje. Snižava
se i torzijska napetost. Smatra se da je energija izvijene kolijevke
za oko 6 kJmol-1 niža od energije konformacije kolijevke pri 25°C.(1,2,3,4)
| Slika
3.
Perspektivni prikaz molekule cikloheksana u konformaciji izvijene
kolijevke. |
|
|
|
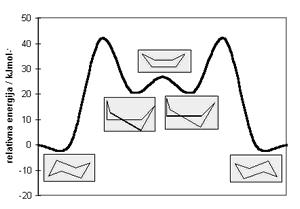
Slika
4.
Energijske barijere uzajamnih pretvorbi konformera cikloheksana.
|
Na Slici 4. zorno se može uočiti kako, pri sobnoj temperaturi,
cikloheksan može brzo prelazi iz jedne konformacije stolice u drugu.
Te su uzajamne pretvorbe posljedica djelomične rotacije oko jednostrukih
C-C veza. Pretpostavlja se da konformacijske inverzije nastaju pomakom
jedne strane oblika stolice pri čemu nastaje izvijena kolijevka;
zatim dolazi do pomaka druge strane izvijene kolijevke, dajući drugu,
odnosno inverznu konformaciju stolice.(2) Analizirajući Tablicu
1. može se zaključiti kako su energijske razlike između pojedinih
konformera cikloheksana primarno diktirane različitim vrijednostima
torzijskih kutova.(3)
|
Slika
5.
Geometrijske pretvorbe konformera cikloheksana.
|
|
Tablica
1. Usporedba geometrijskih
i energijskih parametara konformacijskih izomera cikloheksana.
|
|
konformacija
|
valencijski
kut / °
|
torzijski
kutovi / °
|
DE
/ kJmol-1
|
| |
|
|
|
| planarna |
120
|
0 0 0 0 0 0
|
146
|
| stolica |
112
|
54 -54 54 -54 54 -54
|
0
|
| kolijevka |
112
|
52 0 -52 52 0 -52
|
27
|
| izvijena kolijevka |
112
|
30 -63 30 30 -63 30
|
21
|
Dvanaest vodikovih atoma ne zauzima ekvivalentne položaje u molekuli
cikloheksana. U konformaciji stolice, veze šest vodikovih atoma
usmjerene su okomito na pseudoravninu molekule, a preostalih šest
veza vodikovih atoma usmjerene su malo ispod ili iznad te pseudoravnine.
Veze okomito na pseudoravninu molekule zovu se aksijalne veze, a
one usmjerene u ravnini su ekvatorijalne veze. Tri aksijalne veze
usmjerene prema gore potječu od svakog drugog ugljikova atoma, a
ekvatorijalne veze preostalih triju ugljikovih atoma usmjerene su
također malo prema gore. Atomi vezani preko tih šest veza leže iznad
ravnine molekule. Slični prostorni odnosi vrijede kod triju ekvatorijalnih
i triju aksijalnih veza usmjerenih ispod ravnine molekule. Dakle,
svaki ugljikov atom ima po jednu aksijalnu i ekvatorijalnu vezu
usmjerenu prema suprotnim stranama ravnine molekule (Slika 6.).(2)
|
Slika
6.
Uzajamne pretvorbe ekvatorijalnih (eq) i aksijalnih (ax) vodikovih
atoma u konformaciji stolice cikloheksana.
|
Kad jedna konformacija stolice prijeđe u drugu (inverzija konformacije)
svi ekvatorijalni vodikovi atomi postaju aksijalni, a svi aksijalni
postaju ekvatorijalni. Uzajamna pretvorba ekvatorijalnih i aksijalnih
veza je pri sobnoj temperaturi tako brza, da se svi vodikovi atomi
mogu smatrati ekvivalentnima (Slika 6.).(2)
1. 4. MOLEKULSKA MEHANIKA
Molekulska mehanika jest teorijska (računska) metoda konformacijske
analize. Temelj molekulske mehanike čini klasično poimanje molekule
kao izoliranoga sustava sastavljenoga od atoma, odnosno kuglica,
povezanih kemijskim vezama, odnosno elastičnim oprugama. Sukladno
tome, molekula ima neku vrijednost vlastite potencijalne (konformacijske
ili "naponske") energije uslijed odstupanja od ravnotežne ili "idealne"
geometrije. Položaji atoma u prostoru gdje ta energija ima minimalne
vrijednosti fizički odgovaraju stabilnim konformacijama, tj. određenim
konformerima.(1) Dakle, molekulskom mehanikom želi se kvantitativno
izraziti potencijalna energija molekule koja je funkcijski ovisna
o internim koordinatama. Internim koordinatama se može egzaktno
definirati molekulska konformacija. Uobičajene interne koordinate
jesu; duljina veza (l), nevezna udaljenost (r), valencijski kut
(J) i torzijski kut (j). Međutim, ne postoji strogo ograničenje
broja i vrste internih koordinata, već se one mogu vrlo slobodno
definirati prema potrebama nekog određenog teorijskoga problema.(1)
Ukupna potencijalna (konformacijska) energija molekule (polje sila)
izražava se kao suma elementarnih potencijalnih energija ("elementarnih
napona");(1)
- ˇ vezni potencijal (napon), V(l) - ovisi o duljni veza.
- ˇ nevezni potencijal (Prelog-ov napon), V(r) - ovisi o neveznim
udaljenostima (van der Waals-ove interakcije).
- ˇ potencijal promjene valencijskoga kluta (Bayer-ov ili klasični
napon), V(J) - ovisi o valencijskim kutevima.
- ˇ torzijski potencijal (Pitzer-ov napon), V(j) - ovisi o torzijskim
kutovima
- ˇ elektrostatičke interakcije, V(q) - ovisi o električnim nabojima.
Svaki od navedenih elementarnih potencijala može se prikazati u
analitičkom obliku kao funkcija neke od internih koordinata. Parametri
koji se pojavljuju u tim matematičkim formulama eksperimentalno
su ugodivi, odnosno molekulsko mehanički proračuni se provode na
strogo empirijskoj osnovi. Kako se pronalaze minimumi konformacijske
energije, odnosno kako se optimira geometrija molekule ? Stabilne
konformacije (minimumi konformacijske energije) pronalaze se gradijentnim
iterativnim metodama optimalizacije (minimalizacije). Pod tim imenom
krije se čitav niz matematičkim, numeričkim metoda koje se, orijentirajući
se prema prvim derivacijama energije u nekoj točki, internoj koordinati
(gradijentu) korak po korak (iterativno) sve više približavaju minimumu,
tj. mjestu gdje će sve prve derivacije energije po internim koordinatama
molekule biti jednake nule. Vrijednost energije u točki minimalne
energije (minimumu) određuje termodinamičku stabilnost konformera,
dok razlika energije između minimuma i maksimuma određuje visinu
barijere konformacijskoga prijelaza.(1)
Na koncu valja istaći, kako vrijednosti energija dobivenih molekulsko
mehaničkim proračunima nisu usporedive s eksperimentalnim vrijednostima
na apsolutnoj, već isključivo na relativnoj ljestvici.
|
 |
|
Kao rezultat postojanja aksijalnih i ekvatorijalnih C-H veza u
molekuli cikloheksana u konformaciji stolice postoje dva konformera
monosupstituiranih cikloheksana, npr. konformer klorcikloheksana
sa supstituentom u aksijalnom položaju i konformer sa supstituentom
u ekvatorijalnom položaju. Opća je poznata i prihvaćena činjenica
o mnogo termodinamički povoljnijem ekvatorijalnom položaju supstitucije
u usporedbi s aksijalnim. Prema tome, osnovna ideja i cilj ovoga
rada jest kvantitativno dokazivanje te kvalitativne "udžbeničke"
činjenice pomoću teorijskih, molekulsko mehaničkih proračuna. Sustavnim
mijenjanjem supstituenata različitih steričkih i elektronskih svojstava
ispitivan je utjecaj stereoelektronskih čimbenika na stabilnost
monosupstituiranih cikloheksana, Također, iako postoje eksperimentalne
vrijednosti, dobivene termodinamičkim mjerenjima, energijskih razlika
između ekvatorijalnoga i aksijalnoga položaja supstitucije, ovim
radom nastojala se ispitati pouzdanost i reproducibilnost molekulsko
mehaničkih proračuna u predviđanju i simuliranju eksperimentalnih
podataka.
Molekulsko mehanički proračuni provedeni su MMX poljem sila (5,6).
Grafičke manipulacije molekula načinjene su programom ALCHEMY III
(7). Ovi programi su instalirani na IBM kompatibilnom PC-Pentium/166
računalu.
Minimalizacija konformacijskoga potencijala ispitivanih molekula
provedena je uobičajenom iterativnom metodom najstrmijega spusta
(engl. steepest descent), uz gradijentni kriterij za prestanak minimalizacije
(kada se vrijednost gradijenta spustila ispod programom definirane
vrijednosti, minimalizacija je prekinuta). Svi su proračuni načinjeni
u vakuumu (dielektrička konstanta, e = 1.5), tako da su zanemarene
međumolekulske interakcije, kao i utjecaj otapala. Kao rezultat
molekulsko mehaničkoga proračuna dobivena je vrijednost entalpije
nastajanja (Df H) za ispitivanu konformaciju. Dakle, molekule cikloheksana
i njegovih monosupstituiranih derivata promatrane su kao izolirani
sustavi, a dobivene vrijednosti Df H korištene su za ispitivanje
steroelektronksih utjecaja supstituenata na stabilnost konformacije
monosupstituiranih cikloheksana.
Početna struktura odnosno konformacija cikloheksana dobivena je
običnom geometrijskom konstrukcijom. Geometrija tako "nacrtane"
molekule preliminarno je optimirana MMX poljem sila. Preliminarno
optimiranje nesupstituiranoga cikloheksana rezultiralo je konformacijom
stolice. Upravo zbog te činjenice svi daljnji proračuni rađeni su
na konformaciji stolice, kao najstabilnijoj konformaciji cikloheksana.
Nakon toga, a u cilju konformacijske analize monosupstituiranih
cikloheksana, pojedinačno su dodavani supstituenti najprije u aksijalni,
a potom u ekvatorijalni položaj. Dodavanjem supstituenata očuvana
je tetraedarska sp3 konfiguracija cikloheksanskoga ugljika. Geometrije
aksijalnih i ekvatorijalnih koformera monosupstituiranih cikloheksana
zasebno su optimirane MMX poljem sila. Prije dodavanja svakog novog
supstituenta optimirana je početna geometrija nesupstituiranoga
cikloheksana u konformaciji stolice. Dobivene entalpije nastajanja
aksijalnih i ekvatorijlanih izomera monosupstituiranih cikloheksana
prikazani su u Tablici 2.
|
 |
|
Utjecaj supstitucije na termodinamičku stabilnost aksijalnoga ili
ekvatorijalnoga konformera monosupstituiranoga cikloheksana ispitivan
je s 24 različita supstituenta. Izabrani supstituenti mogu se po
svojim kemijskim značajkama podijeliti u pet osnovnih skupina;
[1] halogeni supstituenti -F, -Cl, -Br i -I
[2] supstituenti kao kisikovi spojevi alkoholi (-OH) i eteri
(-OMe, -OEt i -OBz)
[3] supstituenti kao amini -NH2, -NMe2 i -NMe3+
[4] supstituenti kao karboksilni spojevi karboksilne kiseline
(-COOH), esteri (-COOMe i COOEt) i nitrili (-CN)
[5] alkilni i arilni supstituenti alkili (-Me, -Et, -i-Pr,
-n-Pr, -n-Bu, -tert-Bu, -neopentil, -cikloheksil) i aril (-Ph)
U Tablici 2. jasno se uočava kako su vrijednosti svih relativnih
entalpija nastajanja pozitivne. Obzirom na definiranje relativne
entalpije nastajanja, kao razlike u entalpiji nastajanja aksijalnoga
i ekvatorijalnoga konformera, može se nedvojbeno zajključiti kako
je ekvatorijalni konformer termodinamički stabilniji. Također, vrijednosti
tih relativnih entalpija nastajanja značajno variraju ovisno o naravi
supstituenta.
| Tablica
2. Usporedba
razlika u entalpiji nastajanja za aksijalni i ekvatorijlalni
izomer monosupstituiranoga cikloheksana. |
|
supstituent
|
D
(D f H)mm a, b
|
D
(D f H)exp a,c
|
|
-F
|
0.67
|
0.800
|
|
-Cl
|
1.79
|
1.700
|
|
-Br
|
1.98
|
2.100
|
|
-I
|
0.78
|
1.700
|
|
|
|
|
|
-OH
|
1.42
|
1.300
|
|
-OMe
|
2.42
|
2.90
|
|
-OEt
|
3.03
|
3.80
|
|
-OBz
|
5.54
|
4.20
|
|
|
|
|
|
-NH2
|
7.07
|
5.00
|
|
-NMe2
|
8.34
|
8.80
|
|
-NMe3+
|
16.59
|
17.00
|
|
|
|
|
|
-COOH
|
10.23
|
5.00
|
|
-COOMe
|
9.03
|
4.60
|
|
-COOEt
|
10.83
|
4.60
|
|
-CN
|
0.92
|
0.80
|
|
|
|
|
|
-Me
|
7.44
|
7.10
|
|
-Et
|
7.60
|
7.60
|
|
-i-Pr
|
11.16
|
8.80
|
|
-n-Pr
|
10.75
|
8.80
|
|
-n-Bu
|
9.80
|
8.80
|
|
-tert-Bu
|
20.80
|
24.00
|
|
-neopentil
|
9.36
|
8.40
|
|
-cikloheksil
|
8.22
|
9.2
|
|
-Ph
|
12.94
|
13.00
|
|
|
|
|
|
Legenda: a
Relativna entalpija nastajanja
D
(D f
H) = D
(D f
H)ax - D
(D f
H)eq .
Vrijednosti su izražene u kJmol-1.
b relativna entalpija nastajanje dobivena molekulsko
mehaničkim proračunima
c relativna entalpija nastajanjad dobivena termodinamičkim
mjerenjima.(3)
|
RASPRAVA
Iz
Tablice 2. mogu se izvesti slijedeći zaključci;
halogeni supstituenti
Relativne entalpije nastajanja svih
halogenih supstituentata dobivene molekulsko mehaničkim proračunima
nalaze se u rasponu od oko 1.5 kJmol-1. Kako
veličina halogenog elementa raste (raste vrijednost van der Waals-ovog
radijusa) povećava se i duljina veze C-Hal, a posljedično tome povećava
se i nevezna udaljenost aksijalnoga halogena i aksijalnoga vodika.
Dakle, povećava se termodinamička stabilnost aksijalnoga konformera,
u odnosu na ekvatorijlalni, rezultirajući približno jednakim vrijednostima
relativnih entalpija nastajanja.
supstituenti kao kisikovi i karboksilni spojevi.
Hidroksilna skupina (-OH) pokazuje vrijednosti
vrlo slične kloru, a cijano (-CN) skupina vrijednosti slične fluoru.
Eterski i esterski supstituenti imaju uzajamnno slične vrijednosti,
jer do grananja supstituenata dolazi tek na drugom atomu, u odnosu
na cikloheksanski prsten. Dakle,
promjene vrste eterskih ili esterskih supstituentata imaju maleni
utjecaj na vrijednost relativne entalpije nastajanja. Benziloksi
(-OBz) supstituent, zbog svoje veličine i naglašenije težnje za
stabilnijim ekvatorijalnim konformerom, malo odstupa od navedene
"eterske pravilnosti".
supstituentu kao amini, te alkilni ili arilni supstituent
Sve dok prvi atom ima barem jedan atom vodika ili nepodijeljeni
elektronski par (amini) vezan za sebe, utjecaj promjene supstituenata
na odnos stabilnosti ekvatorijalnoga konformera u odnosu na aksijalni
je nezamjetan. Međutim, čim supstituent posjeduje a
-Me skupinu, ona ulazi u znatne steričke repulzivne (odbojne) interakcije
uzrokujući znatno povećanje relativne entalpije nastajanja (vrijednost
-tert-Bu s a
-Me skupinom je za oko 10 kJmol-1 veća od vrijednosi -neopentila).
Cikloheksilni i fenilni supstituent se, zbog svoje veličine, ponašaju
analogno -OBz supstituentu
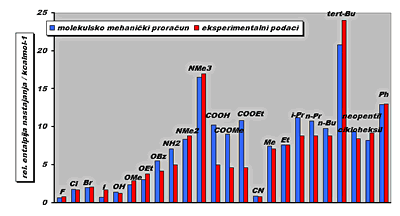 |
Slika
7. Usporedba
vrijednosti relativne entalpije nastajanja dobivenih
molekulsko mehaničkim proračunima s eksperimentalnim
podacima.(3) |
Usporedbom teorijskih (molekulsko-mehaniki
proračuni) i eksperimen-
talnih vrijednosti (Slika 7.) opaža se njihovo razmjerno dobro podudaranje.
Jedino naglašenije odstupanje je uočeno za esterske i karboksilne
supstituente (-COOH, -COOMe i -COOEt). Naime, molekulska mehanika
loše tretira esterski kisik, odnoso karboksilni kisik, u svojim
proračunima što dovodi do precijenjivanja vrijednosti
relativnih entalpija nastajanja.
|
 |
|
Obzirom da je nevezna udaljenost
aksijalnih supstituenata i susjednih aksijalnih vodika uvijek manja
od nevezne udaljenosti ekvatorijalnih supstituenata i njima susjednih
vodika, van der Waals-ove, nevezne repulzive
(odbojne) sile diktiraju termodinamičku stabilnost ekvatorijalnih
konformera. Dakle, molekulsko mehanički proračuni su pokazali da
na termodinamičku stabilnost monosupstituiranih cikloheksana utječu
isključivo sterička, a ne elektronska svojstva
supstituenata.
Također, pokazana je značajna
podudarnost i pouzdanost, kao i reproducibilnost molekulsko mehaničkih
proračuna, unutar granica računskih i eksperimentalnih pogrešaka,
s eksperimentalnim termodinamičkim mjerenjima.
Na koncu se može zaključiti,
kako molekulsko mehanički proračuni mogu poslužiti, kao vrlo brza
i pouzdana metoda, za predviđanje konformacijskih svojstava molekula,
odnosno njihovih geometrijskih i energijskih parametara.
|

 N.
Raos, Konformacijska analiza, Školska knjiga, Zagreb
(1988) 9-17; N.
Raos, Konformacijska analiza, Školska knjiga, Zagreb
(1988) 9-17;
29-33; 41-51.
S.
H. Pine, Organska kemija, Školska knjiga, Zagreb (1994)
132-182.
J.
Dale, Stereochemistry and Conformational Analysis,
Universitetsforlaget & Verlag Chemie, Oslo & New
York-Weinheim
(1978) 147-156.
V.
M. Potapov, Stereochemistry, MIR Publishers, Moscow (1978)
322-333.
J.
J. Gajewski, E. K. Gilbert, J. Mckelvey, MMX An Enhanced
Version of
MM2; in Advances in Molecular Modeling 2, Editor D. Liotta,
Jar
Press, Greenwich, USA (1989).
N.
L. Allinger, J. Am. Chem. Soc. 99 (1977) 8127.
ALCHEMY
III, Tripos Associates Inc., St. Louis, USA (1992).
Istraživanje je provedeno u Zavodu za opću i anorgansku kemiju
Prirodoslovno-matematičkoga fakulteta Sveučilišta u Zagrebu tijekom
ožujka mjeseca 1998. godine.
|
|

|
|

|